but in reality, it utilizes only 12 cores. I ran the command again but with -j 12 but then it utilized around 8. Tried to run it then with 8 cores, but then again the process effectively uses fewer cores. I was wondering if the tag -j has any limits/restrictions? My reference for parallelization is Parallel processing with command-line tasks — LSST Science Pipelines
Any other reference that you think could help clear this problem, I will appreciate a lot
Thanks a lot!
there are no explicit restrictions on how many processes can run with -j option, but there may be bottlenecks due to data dependencies between tasks or other factors that limit concurrency. If pipeline includes many processing steps that depend on each other then later steps need to wait until previous ones finish. And there are additional variables to this of course, some tasks may be waiting on reading/writing to disk instead of using CPU at 100% capacity. How do you measure your concurrency, do you check the number of active processes or CPU utilization? For better diagnostic the log file from your pipetask execution may be helpful.
Hi Andy,
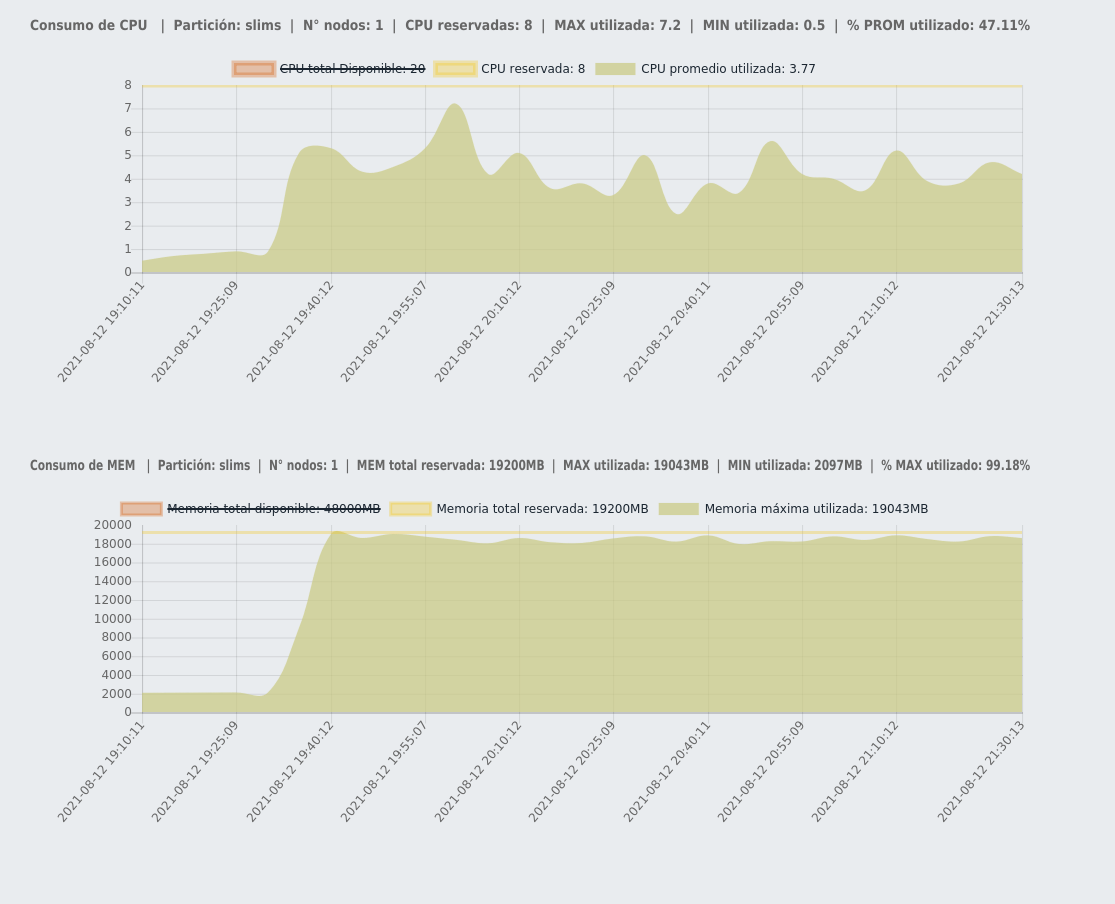
yes, for example In the image below I have the number of cores utilized over time, and in the plot below I have the total memory utilized over time (the labels are in Spanish hehe, sorry). This is for the case of using -j 8. Considering what you say, I will try with a lower number of cores.
Interesting plot. It would still be useful to check ow many pipetask processes are actually executing, could you run something like ps --forest -fu $USER to check the process count while pipetask is executing?
Also I think that pipetask document (How to enable single-node parallel processing) may be confusing. It speaks about spreading processing across multiple CPUs, while what happens in reality is that pipetask tries to execute specified number of processes, but there is no guarantie that they will be able to utilize CPUs with 100% efficiency. Depending on what specific task does the utilization can be lower than 100%.
Ok so, because I’m working with a cluster, usually the process cancels once it evaluates that I’m using less cores than what I configured to use (as seen in the plot that I attached before). So I started a new pipetask with -j 4 and doing what you mention, I get:
Well, that output just shows that pipetask is not running, or at least it is not running on the same host as ps command is running. Does it mean that pipetask was cancelled before you ran ps command or did it run on a different host? I’m sort of lost here, I guess because I’m mostly thinking about how to run pipetask on a local machine but your cluster environment may do things differently? Is there someone with more knowledge of your cluster environment who could help, or is there a documentation for your cluster that explains how things can be controlled?
Yes, the pipetask was cancelled, and then I ran it again with 3 cores -j 3 and then ran ps.
Right, since I’m using a cluster environment, the task is running in cores that are separate from my user, so it kind of makes sense that ps shows as if is not running. I am getting in contact with someone with more knowledge of my cluster environment I will update this post with any progress/improvement.
Thanks a lot for your responses, It really cleared a lot of stuff for me!
It seems that using 4 to 5 cores yields the best results for parallelization with pipetask run (in my case) .
I also heard that there was a tag named --cores that parallelized a task, I was wondering if it still works for the newest version of the LSST Science Pipeline or if it’s now deprecated.
I do not think gen2 version had this --cores option, could it be that this option is something that applies to your cluster environment (which I do not know what it is)? For gen3 the only option that controls concurrency is -j (plus there are few environment variables that control concurrency in math libraries), and this works in almost the same way as in gen2. The main difference is the data dependencies, in gen2 you only run single task on multiple independent data IDs which means there are no dependencies between processes. In gen3 we execute more complicated graphs that usually include multiple processing steps with dependencies between then, which could limit concurrency. I don’t think there is a general advice at this point on how to estimate possible concurrency for a particualar pipeline execution, may need few trials to find an optimum, I guess exactly what you do.
One quick additional comment. If you use the --qgraph-dot FILE option in pipetask qgraph you can visualize the quantum graph (via graphviz) and that should give you a pretty good idea as to how much parallelization you would expect to be possible.
For larger graphs we use a package called ctrl_bps to submit workflows to HTCondor, PanDA, or Pegasus systems (and there is a parsl plugin as well that’s not directly supported by us). We use that for doing the large (>100,000 node) workflows for data release production. ctrl_bps is shipped as part of lsst_distrib.